The challenges of the upcoming exascale supercomputing era in computational biochemistry
Dr. Vedran Miletić (group.miletic.net)
😎 Group for Applications and Services on Exascale Research Infrastructure, Faculty of Informatics and Digital Technologies, University of Rijeka
Research Class, FIDIT, UniRi, 26th January 2022
Stream and recording check
- OBS
- BBB
Dr. Vedran Miletić's previous research work
- Dr. Branko Mikac's group at FER Dept. of Telecommunications
- What to do after finishing the Ph.D. thesis? 🤔
- NVIDIA CUDA Teaching Center (later: GPU Education Center)
- research in Dr. Željko Svedružić’s Biomolecular Structure and Function Group and Group (BioSFGroup, svedruziclab.github.io)
- postdoc in Dr. Frauke Gräter's Molecular Biomechanics (MBM) group at Heidelberg Institute for Theoretical Studies
- collaboration with GROMACS developers from KTH, Max Planck Institute for Biophysical Chemistry (now: Multidisciplinary Sciences), and University of Virginia
RxTx Research
- returned from Heidelberg, became a Senior Lecturer
- 90% working hours teaching (courses + Bura supercomputer), 10% administration, 0% research
- started RxTx Research (rxtxresearch.github.io)
- collaboration with Patrik Nikolić (www.nikoli.ch, former student researcher in BioSFGroup)
- vision: advancing the pharmaceutical drug research by improving the scientific software behind the scenes
- developed open-source high-throughput virtual screening engine RxDock (rxdock.gitlab.io, until promotion to assist. prof.)
Group for Applications and Services on Exascale Research Infrastructure (GASERI)
- The main interest: the application of exascale computing to solve problems in computational biochemistry
- The goal: design better-performing algorithms and offer their implementations for academic and industrial use to
- study the existing molecular systems faster
- study the existing molecular systems in more detail
- study larger molecular systems
Introduction
- a supercomputer is a computer with a high level of performance as compared to a general-purpose computer
- also called high performance computer (HPC)
- measure: floating-point operations per second (FLOPS)
- PC -> teraFLOPS; Bura -> 100 teraFLOPS
- modern HPC -> 1 do 10 petaFLOPS, top 442 petaFLOPS
- future exascalar HPC -> 1+ exaFLOPS
- nearly exponential growth of FLOPS over time (source: Wikimedia Commons File:Supercomputers-history.svg)
{kind=link}

More heterogeneous architectures require complex programming models
- different types of accelerators
- several projects to adjust existing software for the exascale era
- Software for Exascale Computing (SPPEXA)
- Exascale Computing Project (ECP)
- European High-Performance Computing Joint Undertaking (EuropHPC JU)
SPPEXA project GROMEX
- full title: Unified Long-range Electrostatics and Dynamic Protonation for Realistic Biomolecular Simulations on the Exascale
- principal investigators:
- Helmut Grubmüller (Max Planck Institute for Biophysical Chemistry, now Multidisciplinary Sciences)
- Holger Dachsel (Jülich Supercomputing Centre)
- Berk Hess (Stockholm University)
- molecular dynamics visualization: Electron transport chain
GROMEX
The particle mesh Ewald method (PME, currently state of the art in molecular simulation) does not scale to large core counts as it suffers from a communication bottleneck, and does not treat titratable sites efficiently.
The fast multipole method (FMM) will enable an efficient calculation of long-range interactions on massively parallel exascale computers, including alternative charge distributions representing various forms of titratable sites.
SPPEXA Projects - Phase 2 (2016 - 2018)
Planned GROMACS developments (1/2)
- heterogeneous parallelism presently uses GPUs, could be expanded to also use DPUs
- custom-silicon Anton 2 supercomputer's hardware and software architecture could be an inspiration
- identification of packets that do not need to be delivered to all receivers and force reductions
- NVIDIA already offers free developer kits to interested parties for similar purposes
Planned GROMACS developments (2/2)
- molecular dynamics simulations are periodic
- simulation box types: cubic, rhombic dodecahedron
- present design and implementation of the fast multipole method only supports cubic boxes
- it is possible to also support rhombic dodecahedron: ~30% less volume => ~30% less computation time per step required
- potentially apply for HrZZ UIP (if announced)
Potential GROMACS developments
- Monte Carlo (Davide Mercadante, University of Auckland)
- many efforts over the years, none with broad acceptance
- should be rethought, and then designed and implemented from scratch with exascale in mind
- polarizable simulations using the classical Drude oscillator model (Justin Lemkul, Virginia Tech)
- should be parallelized for multi-node execution
- other drug design tools such as Random Acceleration Molecular Dynamics (Rebecca Wade, Heidelberg Institute for Theoretical Studies and Daria Kokh, Cancer Registry of Baden-Württemberg)
Interesting developments in the broader computational biochemistry ecosystem
- RDKit
- RxDock 😇
- data science: KNIME
- applied artificial intelligence, machine learning, neural networks, and deep learning
RDKit and RxDock
- RDKit, the open-source chemoinformatics toolkit
- official blog frequently talks about molecular fingerprints
- database cartridge for PostgreSQL offers scalable molecular storage and retrieval
- RxDock predicts binding modes of small molecules to proteins and nucleic acids
- official comparison with rDock shows example videos
- in the late 2021. we submitted the study of 36 million molecules binding to SARS-CoV-2 main protease
KNIME
- analytics platform
- set of Lego-like blocks that can be connected via GUI
- replaces scripting, easy to use for non-programmers
- state of the art of computational biochemistry methods:
AlphaFold
- protein structure != protein sequence
- sequence: 100 EUR and 20 minutes
- structure: O(100 000) EUR and many years
- earlier computational solutions: Folding@home
- enabled by the evolution of GPUs and developments in AI
- Forbes calls it The Most Important Achievement In AI—Ever: 'Critical Assessment of Protein Structure Prediction co-founder and long-time protein folding expert John Moult put the AlphaFold achievement in historical context: "This is the first time a serious scientific problem has been solved by AI."'
Potential development: HTVSDB
- web interface and REST API to a molecular database and molecular docking service
- open-source software so it could be hosted locally by other research groups at other universities
- unique features: molecular recommendation, federation
- based on RDKit, RxDock, and potentially AlphaFold
- long-term evolution on a best-effort basis
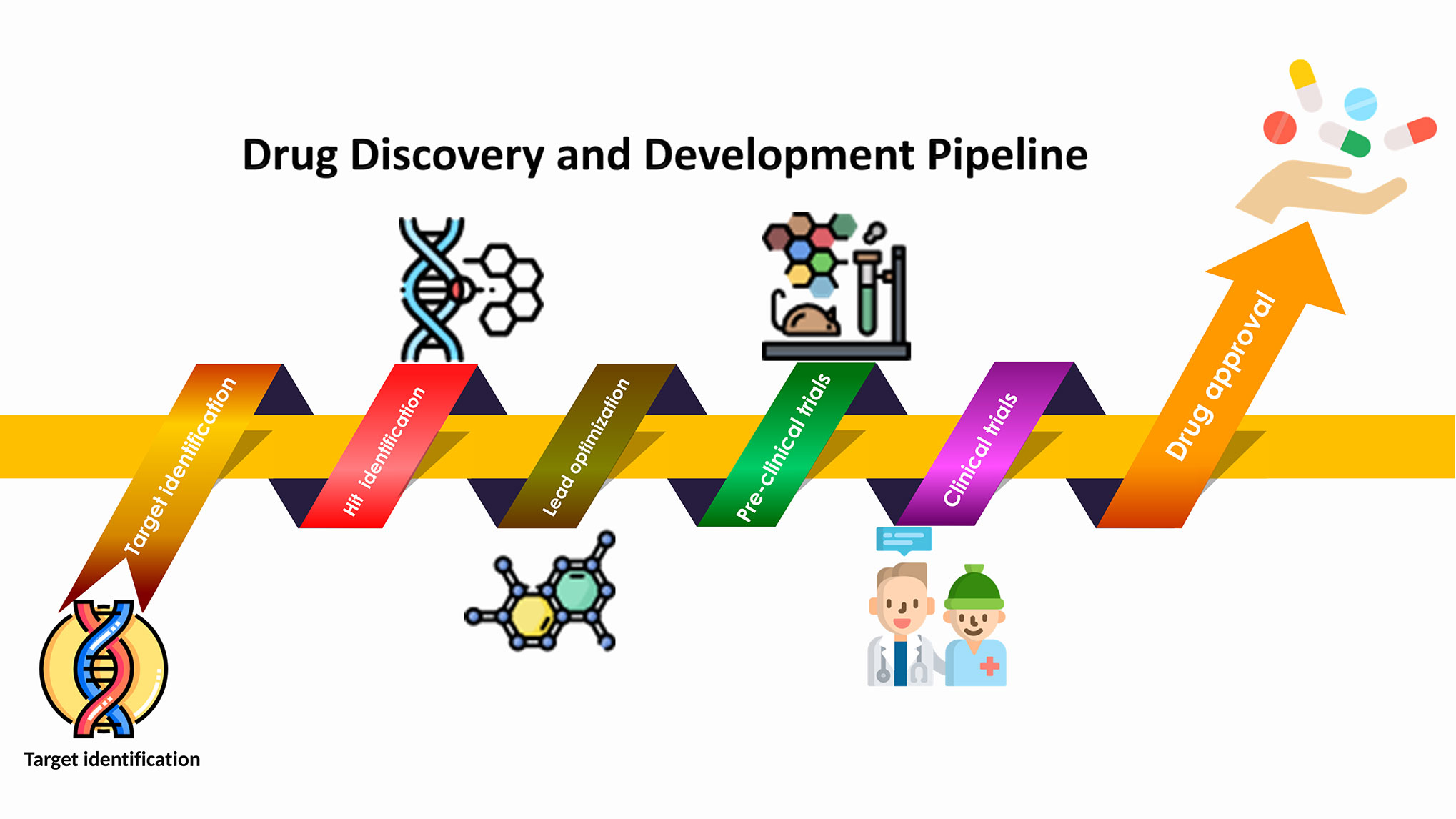
Figure source: Cui W, Aouidate A, Wang S, Yu Q, Li Y and Yuan S (2020) Discovering Anti-Cancer Drugs via Computational Methods. Front. Pharmacol. 11:733. doi: 10.3389/fphar.2020.00733
Unified vision and specific applications
- high-throughput virtual screening and molecular dynamics simulations could be offered as a service to Croatian, regional, and EU research groups
- methods -> algorithms -> applications
- e.g. industry/academic group has a molecular target
- RxDock, RDKit (HTVSDB, KNIME/Python automation): millions of molecules -> tens of molecules
- GROMACS (KNIME/Python automation) -> tens of molecules -> several molecules
Author: Vedran Miletić